www.nebennierentumore.de

Erkrankungen der Nebenniere
Inzidentalom
Unter einem Inzidentalom versteht man eine Raumforderung der Nebenniere, die zufällig bei Schnittbilduntersuchungen aus anderer Indikation in bis zu 8% der Fälle entdeckt wird. Inzidentalome sind häufiger bei älteren Patienten mit Diabetes mellitus, Adipositas und arterieller Hypertonie zu finden. Die Herausforderung beim Inzidentalom liegt in der Differenzierung von primären Nebennierentumoren, bzw. funktioneller von nicht-funktionellen Tumoren, und Metastasen. Dabei ist ein diagnostisches Ziel, ein Phäochromozytom oder ein Kortisol- bzw. Aldosteronproduzierendes Nebennierenadenom von nichtfunktionellen Tumoren zu differenzieren. Die Dignität lässt sich nur bedingt zuverlässig aus der Tumorgröße ableiten. Bei Tumoren mit einem Durchmesser über 6 cm ist wegen des Malignitätsrisikos eine Operation indiziert. Bei kleineren, endokrin inaktiven Tumoren ist eine Verlaufskontrolle gerechtfertigt (nach 6 - 12 Monaten).
Conn-Syndrom
Das Syndrom des primären Hyperaldosteronismus ist klinisch durch die Symptome Hypertension und
Hypokaliämie charakterisiert. Sie sind Folge einer erhöhten adrenalen Sekretion von Aldosteron
und konsekutiver Suppression der Plasmareninaktivität. Weitere charakteristische Symptome
(Muskelschwäche, Muskelkrämpfe, intermittierende Lähmungen, Kopfschmerzen, Polydipsie, Polyurie)
sind überwiegend verursacht durch die Hypokaliämie. Häufigste Ursache des Conn-Syndroms ist ein
unilaterales aldosteronproduzierendes Nebennierenrindenadenom Angiotensin-II-unabhängig bzw. -abhängig),
extrem selten ein aldosteronproduzierendes Nebennierenrindenkarzinom. In etwa 20-40% der Fälle liegt
dem primären Hyperaldosteronismus eine bilaterale mikronoduläre Hyperplasie der Zona glomerulosa
(idiopathischer primärer Aldosteronismus IHA), eine primär makronoduläre Hyperplasie (PMH) oder ein
Glukokortikoid-supprimierbarer Hyperaldosteronismus (GSH) zugrunde. Die Diagnostik soll zunächst den
biochemischen Nachweis der Erkrankung erbringen und anschließend eine Differenzierung zwischen den
verschiedenen Subtypen ermöglichen. Durch weitere Diagnostik sollte das unilaterale Adenom von den
verschiedenen bilateralen Erkrankungen differenziert werden, da nur von einer operation profitieren.
Als sensitivster biochemischer Test gilt die Bestimmung des Aldosteron/Plasmareninaktivitäts-Quotienten.
In frühen Phasen des Conn-Syndroms (klinisch lediglich Erhöhung des Blutdrucks) sind Aldosteron und
Plasmareninaktivität noch im Normbereich, der Quotient zwischen beiden ist allerdings bereits bereits
verändert. Die Diagnose eines primären Hyperaldosteronismus wird dann durch den Fludrokortison-Suppressionstest
bestätigt. Die Differenzierung zwischen unilateraler und bilateraler Erkrankung erfolgt dienen der Orthostasetest
und die selektive Stufenkatheterisierung. Der invasiven Diagnostik geht in der Regel eine Computertomographie
voran, der mikronoduläre Formen noch immer entgehen können.
Präoperativ sollte bei ausgeprägter Hypokaliämie eine Therapie mit dem Aldosteronrezeptorantagonisten
Spironolacton begonnen werden. Zusätzlich ist meistens die Gabe von Kalium und weiteren Antihypertensiva
erforderlich.
Als operatives Standardverfahren hat sich die laparoskopische Adrenalektomie etabliert. Auch ein
retroperitoneoskopisches Vorgehen ist möglich. Aufgrund der Rarität maligner Aldosteronome ist eine totale
unilaterale Adrenalektomie nur selten erforderlich, bei unauffälliger Restnebenniere ist die subtotale
Adrenalektomie ausreichend.
Postoperativ kommt es beim unilateralen primären Hyperaldosteronismus in über 70% der Fälle zu einer schnellen
und dauerhaften Blutdrucknormalisierung. Bei einer bilateralen Erkrankung nimmt die Blutdruckregulation bis zu
einem Jahr in Anspruch und die Rate persistierender Hypertonien liegt höher.
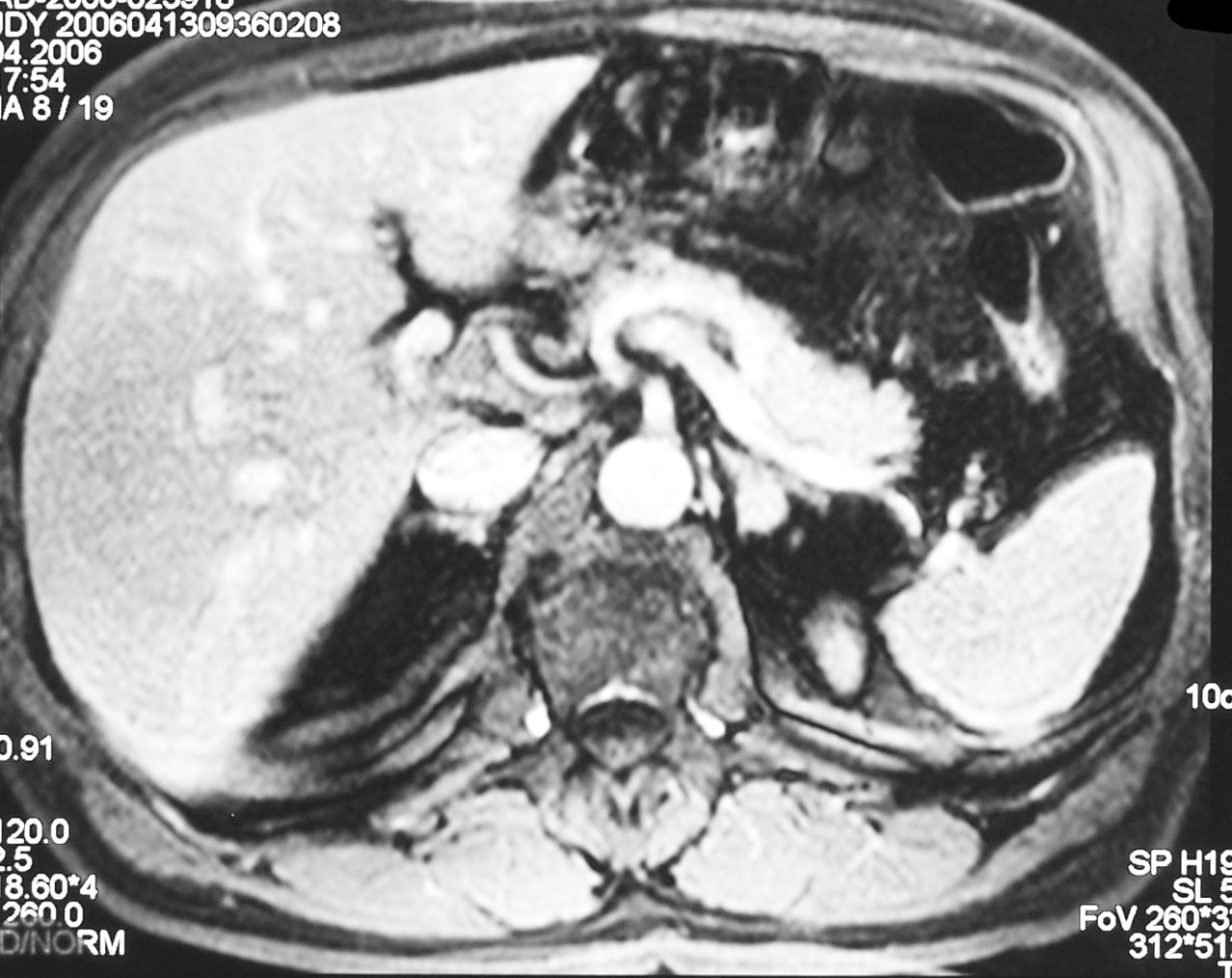
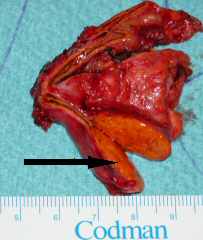
CT und Präparat beim Conn-Syndrom (Pfeil markiert Tumor)
Adrenales Cushing-Syndrom
Das Cushing-Syndrom beschreibt die klinischen und metabolischen Veränderungen unabhängig von der zugrunde
liegenden Ursache. Die häufigste Ursache des Cushing-Syndroms ist die iatrogene Steroidapplikation. 85% der
Fälle zeigen eine bilaterale adrenale Hyperplasie, die meist durch ein ACTH-produzierendes Hypophysenadenom
verursacht werden. Der Nachweis von Hypophysenadenomen erfolgt vorzugsweise mit Hilfe der Kernspintomographie
(MRT). Falls hierdurch eine Lokalisation nicht gelingt (z.B. Mikroadenom) ist die Indikation zur Durchführung
einer simultanen, selektiven, seitengetrennten venösen Blutentnahme aus dem Sinus petrosus inferior gegeben.
In ca. 5-10% der Fälle findet sich eine ektope ACTH-Produktion z.B. durch Bronchuskarzinoide, Thymuskarzinoide,
medulläre Schilddrüsenkarzinome, Phäochromozytome und Inselzelltumoren des Pankreas.
Das adrenale Cushing-Syndrom (10% der Fälle) ist selten und entsteht häufig als Manifestation eines benignen
adrenalen Adenoms oder eines Nebennierenrindenkarzinoms. Als weitere Ursachen sind die mikro- oder makronoduläre
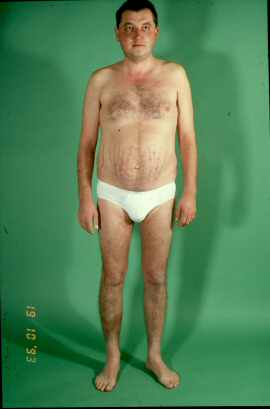
bilaterale Hyperplasie zu nennen. Klinisch bestehen die typischen Symptome des Cushing-Syndroms mit stammbetonter
Adipositas, charakteristische Hautveränderungen, Myopathie, arterielle Hypertonie und Zyklusstörungen.
Zunächst wird das Cushing-Syndrom mit dem Dexamethason-Hemmtest und der Bestimmung des freien Kortisols im
24-Stunden-Sammelurin biochemisch nachgewiesen. Der adrenale Ursprung lässt sich durch ein supprimiertes
Plasma-ACTH und fehlende Stimulierbarkeit durch CRH beweisen. Die Computertomographie ermöglicht die
Differenzierung zwischen uni- und bilateraler Erkrankung.
Bei einseitigem Adenom oder Karzinom besteht die Therapie in der unilateralen laparoskopischen Adrenalektomie.
Die primären adrenalen Hyperplasie erfordern eine bilaterale Adrenalektomie und konsekutiv lebenslange
Substitution mit Glukokortikoiden und Mineralkortikoiden. Nebenniereneingriffe wegen eines Cushing-Syndroms
sind mit einer erhöhten Morbidität belastet. Es ist präoperativ erforderlich, die metabolischen und
kardiovaskulären Störungen der Patienten soweit wie möglich zu beheben. Beim Vorliegen eines schweren
Hyperkortisolismus kann eine Vorbehandlung mit Ketoconazol (Inhibitor der Steroidsynthese) erwogen werden.
Perioperativ ist eine adäquate Thrombose (erhöhter Faktor-VIII-Spiegel)- und Antibiotikapropylaxe
(Vorbeugung postoperativer Infektionen durch den Hyperkortisolismus) unerlässlich.
Phäochromozytom
Phäochromozytome (PZ) sind die häufigsten Tumoren des Nebennierenmarkes (Inzidenz 1-2 pro 1 Mio. Einwohner und Jahr). Phäochromozytome sind Tumoren, die aus den chromaffinen Zellen des Nebennierenmarks entstehen. Sie bilden, speichern und sezernieren Katecholamine. Entsprechend der embryonalgeschichtlichen Entwicklung können auch sog. extaradrenale Phäochromozytome (auch Paragangliome genannt) auftreten, also Tumoren, die in den extraadrenalen Paraganglien entstehen. Das Phäochromozytom wurde früher auch als "10% Tumor" bezeichnet: 10% der Tumoren sind maligne, 10% treten bilateral auf, 10% entstehen extraadrenal und 10% kommen familär vor. Nach neueren Studien wird der Anteil der genetisch bedingten PZ auf 25% geschätzt. Es gibt mehrere hereditäre Tumorsyndrome, die mit Phäochromozytomen assoziiert sind:
| Heredit. Tumorsyndrom | Organmanifestation | Gen |
|---|---|---|
| MEN 2A | Medulläres Schilddrüsenkarzinom Hyperparathyreoidismus |
RET |
| Von-Hippel-Lindau | Hämangioblastome (Retina und ZNS) Nierenzellkarzinom Pankreastumoren |
VHL |
| Neurofibromatose Typ1 | Fibrome an Haut und Schleimhäuten Cafe-au-lait-Flecken |
NF1 |
| Paragangliom-Syndrom | Glomustumoren, Abd. und thorakale Paragangliome | SDHB, SDHD |
Die klinischen Symptome sind vielfältig, häufig unspezifisch und v.a. Folgen der
gesteigerten Katecholaminfreisetzung: Hypertonie (kontinuierlich od. paroxysmal),
Kopfschmerzen, Schwitzen, Palpitationen, Blässe, Gewichtsverlust, Hyperglykämie, Übelkeit,
psychische Beeinträchtigung, orthostatische Dysregulation. Auslöser paroxysmaler
Blutdruckkrisen können körperliche Anstrengung, Manipulation am Tumor, Narkoseeinleitung
und viele chemische Substanzen und Medikamente (Kontrastmittel, trizyklische Antidpressiva, etc.) sein.
Erste diagnostische Maßnahme bei V.a. ein Phäochromozytom sollte immer die Bestimmung der Katecholamine
im Urin sein. Bei betroffenen Patienten beträgt die gemessene Katecholaminkonzentration meistens das
2- bis 40-fache der Norm. Erhöhungen der Katecholaminkonzentrationen bis zum Doppelten des
Normalwertes sind Folge psychischer und physischer Belastung und bedürfen lediglich einer
Verlaufskontrolle. Die höchste Sensitivität hat die chromatographische Bestimmung plasmafreier
Metanephrine und die Bestimmung von Metanephrin und Normetanephrin im 24-Stunden-Sammelurin.
Ist die biochemische Diagnose gestellt, sollte ein ?-Blockade begonnen werden. Zur Lokalisationdiagnostik
kann eine Computertomographie oder Magnetresonanztomographie durchgeführt werden. Die MIBG-Szintigraphie in
SPECT-Technik ist v.a. bei multilokulären Tumoren den Schnittbildverfahren überlegen. Das 18-F-DOPA-PET
kann als zusätzliche Lokalisationsdiagnostik durchgeführt werden.
Nach ausreichender ?-Blockade (normo- bis hypotoner Blutdruck, orthostatische Dysregulation, keine
ST-Strecken-Veränderungen, VES < 5/min im EKG, "verstopfte" Nase) unterscheidet sich die Anästhesie nicht
von der Narkose bei hormoninaktiven Nebennierentumoren. Standardtherapie des unilateralen sporadischen
Phäochromozytoms ist die minimalinvasive Adrenalektomie. Sowohl die laparoskopische als auch die
retoperitoneoskopische Operation ist möglich. Bei einer bilateralen Erkrankung ist eine minimalinvasive
subtotale Resektion empfehlenswert. Der belassene Parenchymrest verhindert eine Nebenniereninsuffizienz
mit Gefahr der Addison-Krise. Ein konventionelles Vorgehen sollte bei Tumoren mit einem Durchmesser über 8
cm oder radiomorphologisch festgestellten Malignitätskriterien gewählt werden. In unserer Klinik wird der
transabdominelle Zugang über einen Subkostalschnitt bei Tumoren über 8cm Durchmesser bevorzugt. Bei größeren
Tumoren oder deutlichen Malignitätskriterien kann auch ein thorakoabdomineller Zugang gewählt werden, der die
beste Übersicht im Hinblick auf die empfohlene Lymphadenektomie bietet.
Beim Phäochromozytom sollte nach 6 Wochen und 6 Monaten postoperativ nachuntersucht werden (Bestimmmung der
Mtea- und Normetanephrine im Sammleurin). Insgesamt werden Nachuntersuchungen für 10 Jahre empfohlen, bei
Patienten mit Phäochromozytom assoziierten Syndromen und extraadrenalen Gangliomen sogar lebenslänglich.
Nebennierenkarzinom
Nebennierenkarzinome sind selten, meist jedoch sehr maligne Tumoren. Die jährliche Inzidenz liegt bei ca.
1 auf 2 Mio. Einwohner. Die Diagnosestellung erfolgt häufig erst bei fortgeschritteneren Stadien mit
lokaler Invasion oder Fernmetastasierung. Die Prognose der Erkrankung mit einer durchschnittlichen
5-Jahresüberlebensrate von 20% ungünstig.
Grundsätzlich werden hormonaktive und hormoninaktive Karzinome unterschieden. Eine endokrine Symptomatik
besteht nur bei etwa 40% der Betroffenen. Die häufigste endokrine Störung ist die autonome Glukokortikoidsekretion
mit Ausbildung eines Cushing-Syndroms. Androgenbildende Tumoren führen bei Frauen zur Virilisierung.
Östrogenproduzierende Nebennierentumoren sind beim Mann immer als maligne anzusehen. Symptome sind Gynäkomastie,
Hodenatrophie, Oligospermie und Impotenz. Außerdem kann ein erhöhter DHEAS-Wert im Serum auf das Vorliegen eines
Karzinoms hindeuten.
Die Einteilung der Nebennierenkarzinome erfolgt nach der TNM-Klassifikation bzw. nach MacFarlane:
| Stadium | TNM-Klassifikation |
|---|---|
| I | T1 N0 M0 |
| II | T2 N0 M0 |
| III | T3 N0 M0 oder T1-3 N1 M0 |
| IV | T1-3 N0-1 M1 |
Bemerkungen: T1 Tumor<5cm, T2 Tumor>5cm, T3 lokal infiltierend, N1 regionaler LK-Befall, M1 Fernmetastasen;
Wenn bei Patienten Symptome der Raumforderung auffallen, liegt in der Regel initial bildgebende Diagnostik
vor. CT und MRT sind gleichermaßen geeignet. Die 18-Fluordesoxyglukose-Positronenemissionstomographie
(18F-FDG-PET) kann zur Abgrenzung zwischen benignen und malignen Nebennierentumoren hilfreich sein.
Zudem können im Rahmen des Stagings Metastasen sichtbar gemacht werden. Die Metastasensuche sollte eine
Thorax- und Abdomen-CT, eine Konchenszintigraphie und ggf eine 18F-FDG-PET umfassen.
Die klinischen Symptome des Patienten leiten die zielgerichtete endokrine Diagnostik. Aber auch bei
klinisch endokrin inaktiven Tumoren ist eine biochmeische Diagnostik sinnvoll, da eine endokrine
Aktivität durchaus auch subklinisch vorhanden sein kann. Zudem kann so ein geeigneter Tumormarker zu
postoperativen Überwachung identifiziert werden. Vor geplanter Adrenalektomie muss auf jeden Fall ein
subklinisches adrenales Cushing-Syndrom ausgeschlossen werden, um eine postoperative Nebenniereninsuffizienz
zu vermeiden. Außerdem muss ein Phäochromozytom zur Vermeidung intraoperativer Komplikationen ausgeschlossen werden.
| obligat | Serumkortisol im Dexamethasonhemmtest (2mg) DHEA-S Katecholamine im 24-h-Sammelurin |
| Bei entsprechender klinischer Symptomatik | Testosteron, 17-ß-Östradiol, LH, FSH Reninaktivität (-Konzentration), Aldosteron im Plasma, Desoxykortikosteron im Serum |
| Cushing-Syndrom | Kortisol im 24-h- Sammelurin Hochdosierter Dexamehasonhemmtest (8mg) CRH-Stimulationstest |
| Virilisierung / Feminisierung | LHRH-Test DHEA-S, Testosteron, 17-ß-Östradiol im Dexamethasonhemmtest |
| Mineralkortikoidhypertonie | Aldosteron-18-Glukoronid / Tetrahydroaldosteron im 24-h-Sammelurin NaCl-Belastungstest |
Der einzige kurative Ansatz zur Therapie des adrenokortikalen Karzinoms ist radikale chirurgische Entfernung allen Tumorgewebes. Bei allen lokal begrenzten Stadien besteht folglich so gut wie immer eine Operationsindikation. Bei Patienten mit Fernmetastsen sollte aufgrund der infausten Prognose eine Operationsindikation kritisch gestellt werden. In Anbetracht der besten Übersicht wird am häufigsten ein konventioneller thorakoabdomineller Zugang eingesetzt. Die Operation beinhaltet die radikale Entfernung des Primärtumors und in den meisten Zentren eine regionale Lymphadenektomie, wobei jedoch ein prognostischer Vorteil der Lymphknotenentfernung bislang nicht nachgewiesen werden konnte. Die Resektion von Fernmetastasen kann im Einzelfall sinnvoll sein.
Nebennierenhyperplasie
Als Hyperplasie bezeichnet man die Vergrößerung einzelner Struktureinheiten des Organs über die Norm
hinaus. Sie können diffus oder in Form von Knoten auftreten und alle Schichten der Nebenniere betreffen.
Eine Unterscheidung erfolgt in kongenitale und erworbene Hyperplasien.
Die kongenitalen Hyperplasien beruhen auf einem Effekt von Enzymen, die in die Biosynthese der
Kortikoide einbezogen sind. Durch einen Mangel dieser Enzyme (am häufigsten 21-a-Hydroxylase, 11-ß-Hydroxylase,
3-ß-Hydroxysteroiddehydrogenase) kommt es über endokrine Regulationsschleifen zu einer vermehrten Produktion
von ACTH, welches eine vermehrte Bildung von Kortisolvorstufen in den Nebennieren zur Folge hat. Dies führt
zu einer diffusen Hyperplasie der Nebennierenrinde.
Häufigste Form der erworbenen Hyperplasien ist die normaktive mikronoduläre Hyperplasie mit Noduli zwischen
1 - 2 mm im Durchmesser, viele Sektionen zeigen derartige Befunde. Ihre Entstehung beruht auf arteriellen
Durchblutungsstörungen des Nebennierenparenchyms, die dazu führen, dass die mangelhaft durchbluteten Areale
atrophieren, während die gut durchbluteten reaktiv hyperplastisch werden. Typisch sind mikronoduläre
Hyperplasien auch beim hypothalamisch- hypophysär bedingten Morbus Cushing. Das Gewicht der Nebennieren
ist hier meist nur gering bis nicht erhöht. Mit zunehmender Krankheitsdauer kann es aber zu einem Wachstum der
Knoten kommen, sodass aus Mikro- nun Makronoduli (Durchmesser > 2mm) werden.
Ein Übergang in Adenome mit zunehmender funktioneller Autonomie wie bei einem primär adrenalen Cushing- Syndrom
gilt dabei als möglich. So kann aus einem primär hypothalamisch- hypophysären Cushing- Syndrom ein adrenales
Cushing- Syndrom werden.
Nebennierenadenome
Histologisch handelt es sich um eine gutartige Vermehrung von Drüsenzellen der Nebennierenrinde. Makroskopisch stellen sich die Nebennierenadenome fast immer unilateral und solitär dar. Ihr Durchmesser liegt zumeist unter 5cm und ihr Gewicht bei unter 50g, jedoch sind auch Gewichte über 100g möglich. Sie zeigen histologisch eine gelblich- bräunliche Schnittfläche, Nekrosen fehlen meistens. Die Adenomzellen sind meist größer als die normalen Rindenzellen, die Zellkerne sind häufig etwas vergrößert und chromatinreicher. Histologisch können differentialdiagnostische Schwierigkeiten bestehen gegenüber Phäochromozytomen und Nebennierenkarzinomen, insbesondere bei größeren lipidarmen Tumoren mit diffusen Wachstum. Adenome sind mit über 50% aller Tumoren der Nebenniere die am häufigsten diagnostizierten Raumforderungen der Nebenniere. Die tatsächliche Inzidenz von Adenomen lässt sich sehr schwierig determinieren, in verschiedenen Autopsiestudien fand man Nebennierenadenome größer 2 - 5 mm in bis zu 5,7% der Untersuchten, wobei die Inzidenz mit zunehmendem Alter anstieg. Nebennierenadenome können mit verschiedenen endokrinologischen Krankheitsbildern vergesellschaftet sein, die Angaben in der Literatur zur Häufigkeit von hormonsezernierenden Adenomen differieren dabei sehr: 5 - 47% aller Adenome sezernierten Kortisol, 1,6 - 3,3 % produzierten Mineralkortikoide, extrem selten fand man benigne Läsionen der Nebenniere, welche Androgene oder Östrogene sezernierten.

Prof. Dr. D.K. Bartsch